1. Introduction
Colorectal cancer (CRC) is a lethal disease, with about 149,500 new cases expected and about 52,980 estimated death in 2021 in the USA [
1]. Therapies for CRC vary based on the severity of the disease. Surgery is used for most tumors in the early stages of CRC, whereas surgery in combination with anti-cancer drugs is used in later stages [
2]. Due to high mortality rates, metastasis, recurrence, and chemoresistance in later stages of CRC, new therapeutics for CRC are urgently needed.
CRC tumorigenesis is marked by several “driver mutations” of genes encoding different tumor suppressors and promoters, including adenomatous polyposis coli (APC), p53, and Kirsten rat sarcoma viral oncogene homolog (KRAS) [
3,
4,
5]. Among other effects, these mutations promote the activation of the nuclear factor (NF)-κB family of transcription factors, which is critical for a number of processes of tumorigenesis, including cellular proliferation, migration, and control of apoptosis [
6,
7].
There are five NF-κB family members in mammals (RelA (p65), RelB, c-Rel, NF-κB1 (p50), and NF-κB2 (p52)). In particular, “classical” NF-κB signaling is usually mediated by the p65/p50 heterodimer as reviewed by May et al. where it can be activated from its latent form in the cytoplasm and translocated to the nuclear to bind to κB consensus sequences in promoters to trigger target gene expression [
8]. The functions of the proteins encoded by these genes frequently affect inflammation, the cell cycle, cell growth, angiogenesis, and cancer progression [
9]. Unsurprisingly, hyperactivation of the NF-κB pathway is closely related to deregulation of the immune system and cancer and progression, including in CRC [
3,
4,
10,
11,
12]. Thus, novel regulators of NF-κB signaling could have great potential as new therapeutic targets in CRC. In this regard, identification of novel regulators of NF-κB is of particular importance as it may open new avenues for future CRC treatment.
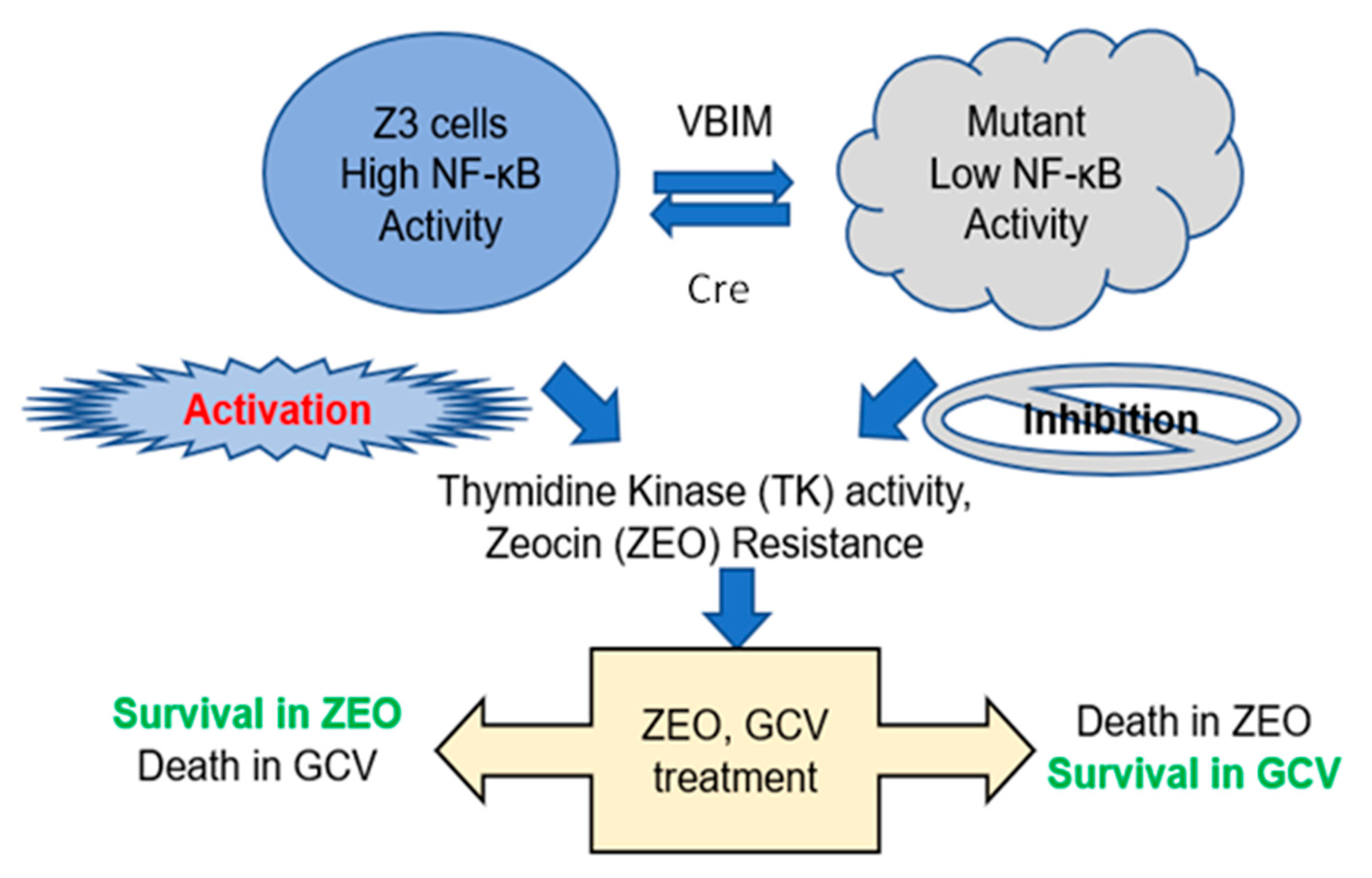
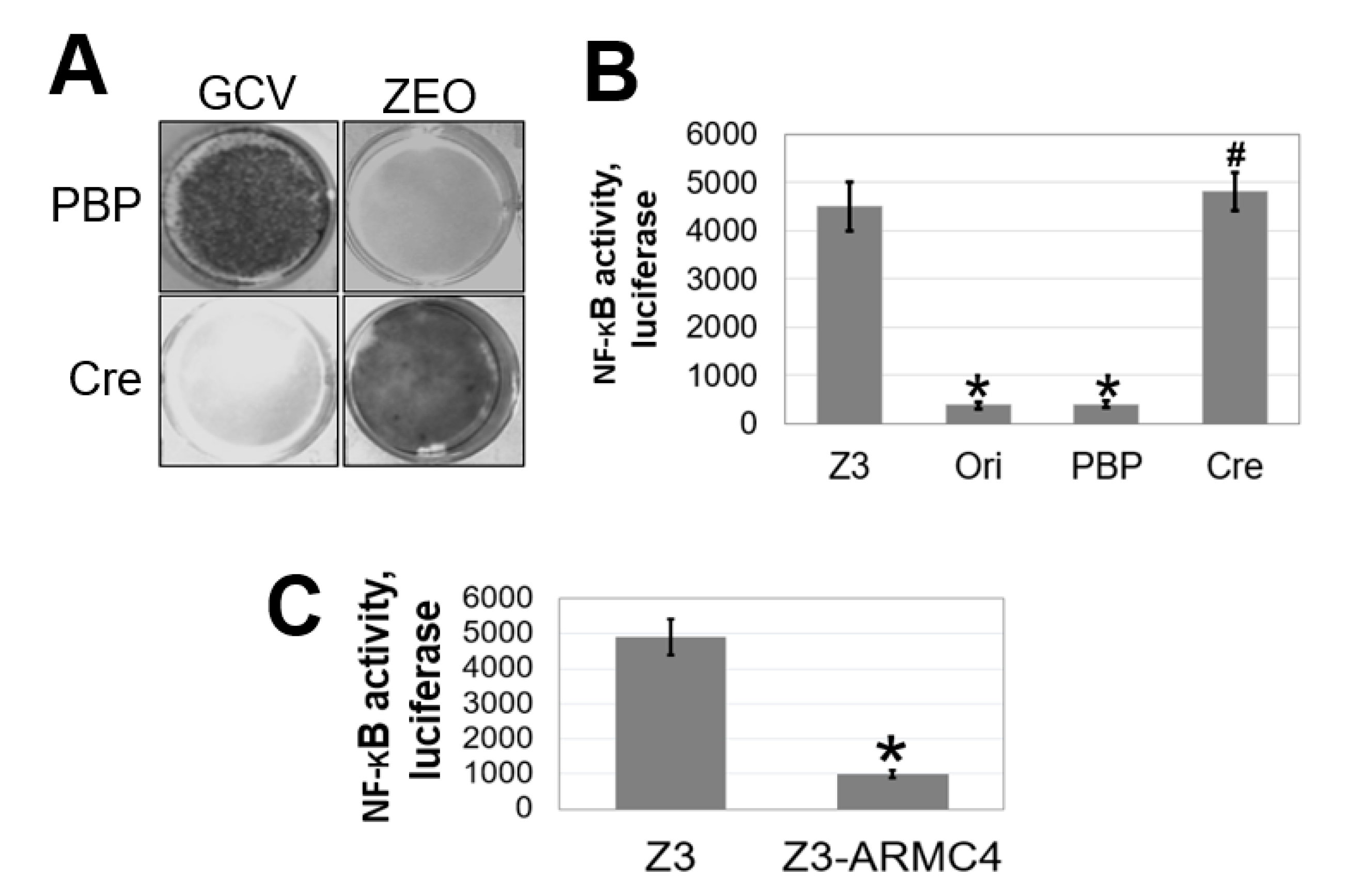
To identify novel therapeutic targets in the NF-κB pathway, we used a robust lentiviral validation-based insertional (VBIM) method to develop mutant cell lines that overexpress specific cellular proteins that may function as NF-κB negative regulators, thus leading to an altered cell phenotype (low NF-κB activity) that is reversible by using the Cre-lox recombination system. A full description of the VBIM technology is presented in our previous publication and has already been used to identify other regulators of NF-κB signaling [
9]. In the current study, we employed this powerful method to identify armadillo repeat-containing protein 4 (ARMC4)/Outer dynein arm docking complex subunit 2 (ODAD2) as a negative regulator of NF-κB. Commonly, this gene is referred to as ARMC4, so it will be presented as ARMC4 hereafter. Prior to the work described here, ARMC4 had been identified as a theoretical therapeutic target only through genome-wide association studies (GWAS) in gastric, ovarian, and breast cancers [
13,
14]. Importantly, ARMC4 is a member of the ARM domain-containing superfamily, which includes APC and β-catenin, both of which are widely known to be dysregulated in CRC and can promote aberrant wingless/integrated (Wnt) pathway signaling [
15]. Despite a plethora of information on other superfamily members, little is known about ARMC4’s regulation and function. Previous knowledge regarding ARMC4 is that it plays a critical role in the rare disorder primary ciliary dyskinesia (PCD) [
16,
17] and mouse spermatogenesis [
18]. In PCD patients, multiple specific point mutations of ARMC4 cause dysfunction in its ability to bind to cilia [
16,
17].
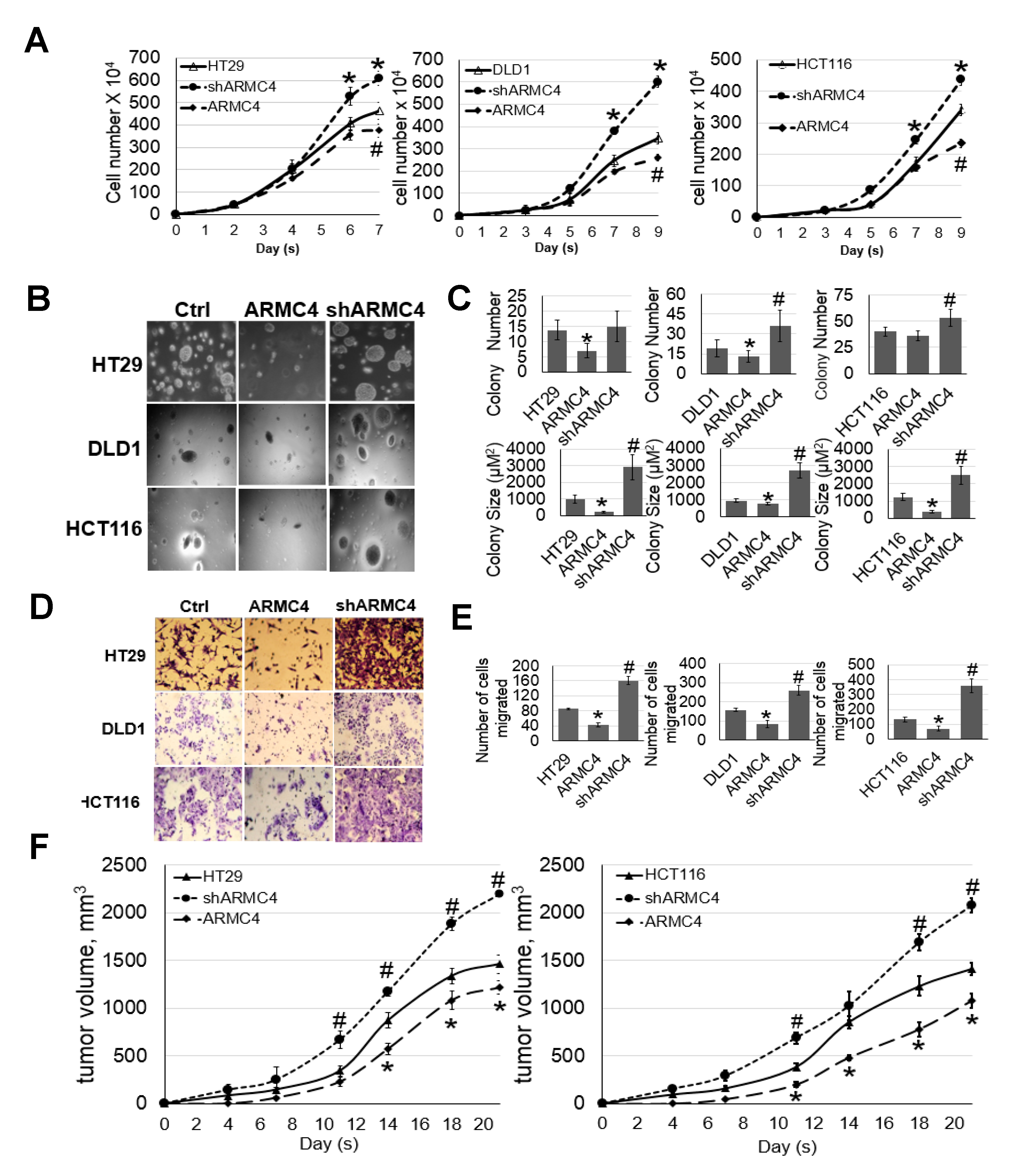
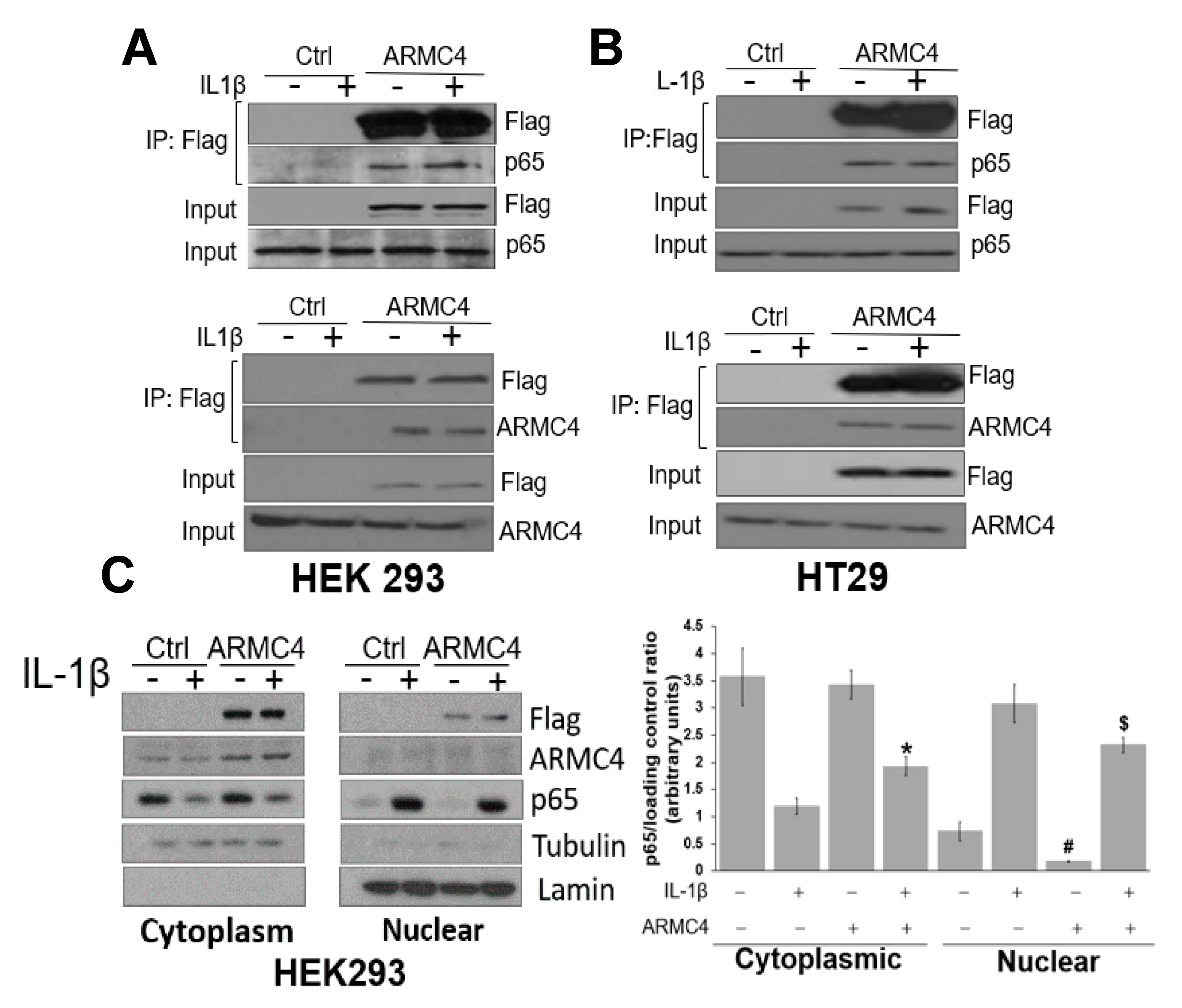
We now find that ARMC4 is a novel negative regulator of NF-κB and also a potential tumor suppressor in CRC. High expression of ARMC4 downregulates the expression of NF-κB-dependent genes, many of which are cancer related. Furthermore, high ARMC4 expression in CRC cells reduces NF-κB activity, cellular proliferation, anchorage-independent growth, and migratory ability in vitro and significantly decreases xenograft tumor growth in vivo, while shARMC4 knockdown has the opposite effect. Moreover, co-immunoprecipitation (co-IP) experiments confirmed that ARMC4 forms a complex with the p65 subunit of NF-κB. Interestingly, immunohistochemistry (IHC) data showed remarkably lower ARMC4 expression levels in CRC tumors than that in normal tissues. Taken together, our data reveal that ARMC4 is a new tumor suppressor in CRC, making it an attractive novel diagnostic biomarker and therapeutic target in this disease.
3. Discussion
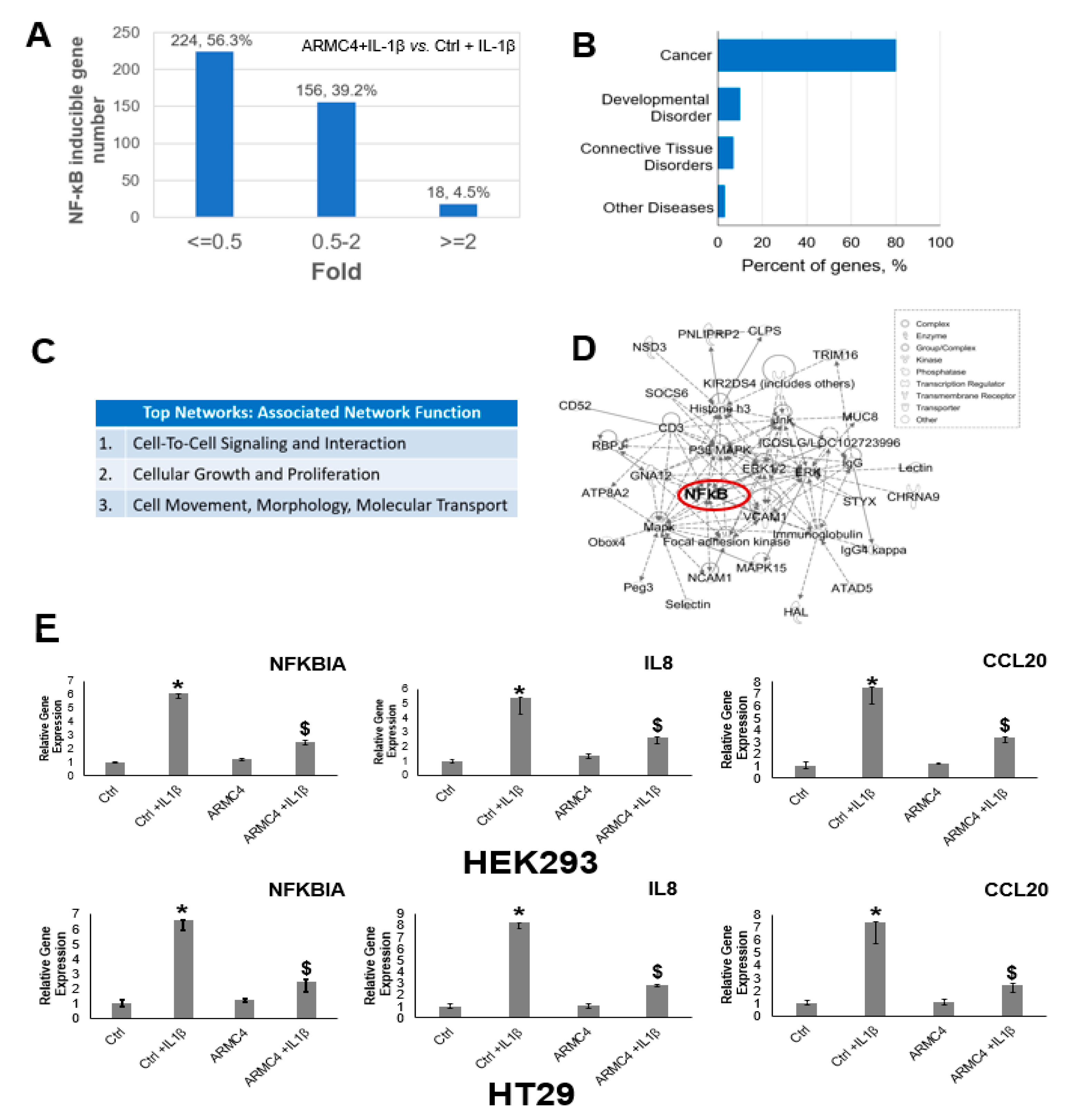
Our work here suggests a previously unknown role of ARMC4 in NF-κB signaling. ARMC4 is newly identified as a novel negative regulator of NF-κB using our VBIM technology (
Figure 1 and
Figure 2). High ARMC4 expression downregulated NF-κB target genes as well as had a marked effect on the regulation of gene networks (
Figure 3 and
Figure 4, and
Supplementary Figure S2). Our data so far suggests ARMC4 may function as a tumor suppressor in CRC. We show that high ARMC4 expression decreased cellular proliferation, migration, and anchorage-independent growth. Additionally, high expression of ARMC4 decreased tumor growth in in vivo NSG mouse models (
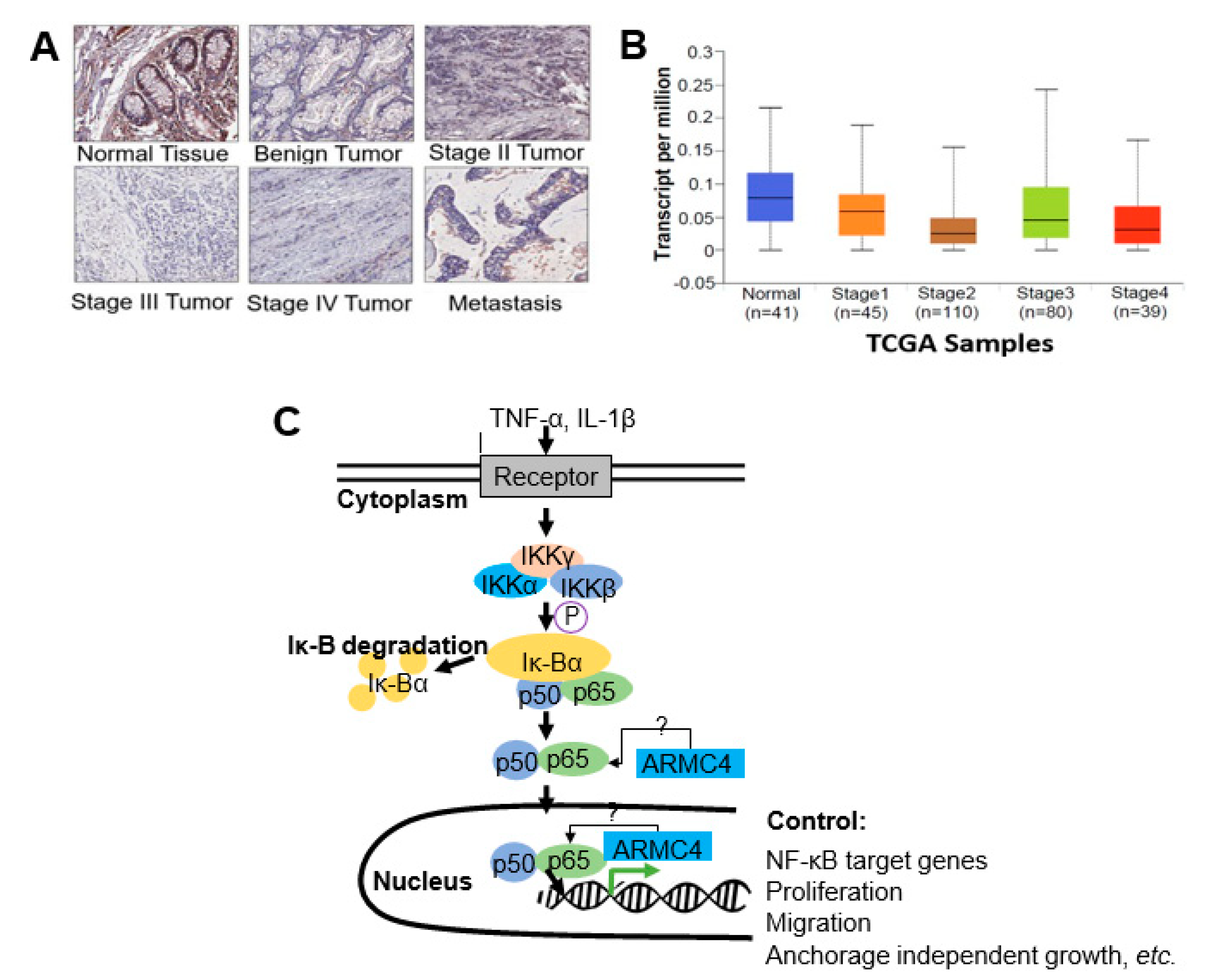
Figure 5). Mechanistically, not much is known about ARMC4’s functions in cancer. Our mechanistic studies suggest ARMC4 and the p65 subunit of NF-κB may complex together. Furthermore, as our cell fractionation studies show, ARMC4 and p65’s interactions may lead to possible sequestration or lagging of p65’s translocation to the nucleus (
Figure 6). Furthermore, we have shown that ARMC4 functions downstream of the classical negative regulator of NF-κB activity, the step of IκBα degradation (
Supplementary Figure S1). ARMC4 expression may also be decreased in CRC patient samples, supporting the notion that ARMC4 is a novel tumor suppressor in CRC (
Figure 7).
While the ARMC4’s novel role in CRC may be clinically relevant, there are still several questions we aim to answer regarding future studies. One major logical question is if ARMC4 is mutated in CRC when compared to normal tissues. Searches of several CRC databases indicate a few missense mutations, which are fairly rare (data not shown). Interestingly, the mutations identified in CRC have never been associated with ARMC4’s known functions in PCD and mouse spermatogenesis.
Another main question we had was whether ARMC4 showed similarities to known tumor suppressors associated with CRC. For example, APC and p53, two well-characterized tumor suppressors, both have shown that their mutations could specifically drive CRC progression. Notably, p53 is mutated in about 70% of CRC patients [
22]. Cooks et al. reported that mutant p53 can cause constitutive NF-κB activity, promote chronic inflammation, and subsequently drive further mutation in the progression of CRC [
23,
24]. Wild-type p53 acts to limit proliferation through transcriptional regulation of a cell’s life cycle by cell cycle arrest, senescence, and apoptosis [
23,
25]. Due to its critical cellular regulatory functions, mutations in p53 can lead to the promotion of cancer and are often seen in CRC [
25]. Another famous tumor suppressor in CRC is APC, whose mutations cause dysregulation of the Wnt/β-catenin pathway, leading to CRC progression. APC is mutated in about 80% of all forms of CRC [
26]. It controls the signaling of the Wnt/β-catenin pathway through cytoplasmic proteasomal degradation of β-catenin [
27,
28]. Loss of this function through mutated APC leads to disruption of apoptosis regulated by the Wnt/β-catenin pathway, leading to CRC progression [
27]. It is noteworthy that while no links have been drawn between ARMC4 and its superfamily members APC and β-catenin, ARMC4 does share several major structural similarities with APC. The ARMC4 protein is made up of 10 tandem armadillo repeat motifs (ARMs), which form into ARM domains and one HEAT repeat, while APC contains 8 ARM domains and 4 coiled coil motifs. The ARM repeats are known to be important for transduction of Wnt signaling in embryonic development and are purported to form a superhelix that mediates protein-protein interactions [
29]. Since both ARMC4 and APC have tandem ARM domains, it is possible that they can both form ARM superhelices, allowing them to bind other proteins. We speculate that the structural relationships between ARMC4 and APC suggest potential similar functional roles for ARMC4 and APC in binding proteins and possibly similar functions as tumor suppressors. However, these possibilities need to be further tested in future studies. While the data we have shown so far suggests several potential mechanisms of interaction between NF-κB and ARMC4, more details remain to be discovered. Of particular interest to us is the structure-function relationship between ARMC4 and APC. So far, we have shown full-length highly expressed ARMC4 decreases NF-κB transactivation activity. Additionally, our search of CRC patients sample databases indicates a low prevalence of mutations in ARMC4 in some CRC patients. Of further interest to us is the recent study by Liang et al., which indicates ARMC4 as a mutated gene in several patients with CRC ranging from stage I to stage III tumors with missense mutations [
30]. This is consistent with our notion that ARMC4 functions as a tumor suppressor in CRC. In the future, we may generate truncated ARMC4 mutants to potentially disrupt the secondary structure of ARMC4 and its interactions with p65, thereby pinpointing ARMC4’s functional domains that are crucial to the regulation of NF-κB activity. Besides these avenues, an additional interesting aspect is the CRC microenvironment. We show that high ARMC4 expression in CRC cells produced the conditioned media that remarkably reduced NF-κB activity in stable 293-NF-κB reporter cells (
Figure 3C). This phenomenon may be due to several reasons, such as ARMC4 downregulating target gene expression such as pro-inflammatory cytokines and thus their release into the microenvironment. To further test this hypothesis, we may pursue future studies such as cytokine array experiments for the CRC conditioned media.
Additionally, we show that ARMC4 has decreased expression in later stages of CRC (
Figure 7), but the reasons behind this phenomenon are unknown. ARMC4 is known to have regulatory roles in PCD and mouse spermatogenesis, but what regulates ARMC4 itself is not known. In future work, we would like to identify regulators of ARMC4. This effort may help us uncover a novel therapeutic avenue for regulating constitutive NF-κB activity in CRC via control of ARMC4 expression. Besides its application as a potential therapeutic target, we may further pursue studies to determine if ARMC4 is a biomarker of CRC progression.
4. Materials and Methods
4.1. Cell Lines and Antibodies
The human embryonic kidney (HEK) 293 cells, also referred to as 293 cells, were cultured in Dulbecco’s modified Eagle’s media (DMEM) supplemented with 100 U/mL penicillin, 100 mg/mL streptomycin, and 10% fetal bovine serum (FBS). HEK293 cells were previously described [
12]. The HT29, DLD1, and HCT116 colon cancer cell lines were purchased from the American Tissue Culture Collection (ATCC) (Manassas, VA, USA) and were cultivated in RPMI1640 media with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal calf serum. Experiments were carried out when the cells reached 90% confluence. The following antibodies were obtained from commercial sources: anti-ARMC4, anti-NF-κB p65, and anti-IκBα were from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Anti-β-actin and anti-Flag were from Sigma-Aldrich (St. Louis, MO, USA).
4.2. Virus Production, Cell Infection, and Selection
The VBIM and pLV-tTR-KRAB-Red lentiviruses were packaged in 293T cells using second-generation packaging constructs pCMV-dR8.74 and pMD2G (both packaging plasmids and pLV-tTR-KRAB-Red were kind gifts of Dr. Didier Trono, University of Geneva, Switzerland. Retroviruses encoding Cre-recombinase or empty vector control were packaged in Phoenix-Ampho cells. Supernatant media-containing virus, collected at 36–48 h, were supplemented with 4 µg/mL polybrene before being frozen in aliquots. To perform selections in Z3 cells, infections were performed so that 70–90% of each population was GFP-positive before selection with drugs. Z3 cells, pretreated with 25 μg/mL of zeocin for 7 days to remove any background mutants, were cultured at 1 × 105 cells/well, into 30 wells of 12-well plates. The next day, the cells were infected with the three different VBIM viruses (SD1, 2, and 3). The medium was replaced 24 h after infection, and the cells from each well were split and transferred into a 15 cm plate 48 h later. After another 24 h, medium containing 0.1 μg/mL of GCV was added and replaced every three days. Individual clones were picked after 2 weeks.
4.3. Construction of Stable ARMC4 Overexpressing or shARMC4 Cells
ARMC4 gene was cloned into the lentiviral vector as a full-length cDNA or shRNA pool (containing five different shRNA constructs) against ARMC4 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Viruses were generated and used to infect HT29, HCT116, DLD1, and HEK293 cells. Since both the lentiviral vector and the shRNA pool carry the puromycin resistance marker, puromycin-resistant clones were selected in 1 μg/mL puromycin after virus infection. Then cells were pooled and tested for the best overexpression and knockdown by the Western blot method with antibodies against ARMC4.
4.4. Transfections and Luciferase Assays
For NF-κB luciferase assays, the κB-luciferase construct p5XIP10 κB (contains five tandem copies of the NF-κB site from the IP10 gene) was transfected transiently into the cells, and luciferase activity was assayed 48 h later. A β-galactosidase (β-gal) construct was co-transfected to normalize for transfection efficiency. The cells were then washed with cold phosphate-buffered saline (PBS) and lysed in 80 μL of 5X lysis buffer from Promega Corporation (Madison, WI, USA). After incubation on ice for 15–20 min, cell debris was pelleted at 15,000× g for 5 min at 4 °C. A measure of either 30 μL of luciferase assay substrate or β-gal substrate (Promega Corporation) was added to 20 μL of supernatant solution before being read in a luminometer. The relative luminescence was normalized to the optical reading of β-gal (Bio-Rad Laboratories, Hercules, CA, USA). Luciferase activity was measured using a Synergy H1 Multi-Mode Reader (BioTek Instruments Inc., Winooski, VT, USA).
4.5. PCR, Quantitative PCR
The total RNA from mutant cells was extracted with the TRIzol
® reagent at room temperature following the protocol from manufacturer (Invitrogen Life Technologies, Waltham, MA, USA). Reverse transcription was performed using oligo DT20 primers with the SuperScript III First-Strand Synthesis System kit and protocol (Invitrogen Life Technologies, Waltham, MA, USA). Standard PCR reactions were further performed by using a forward primer from the VBIM vector [
9] and Oligo dT primer from the SuperScript III First-Strand Synthesis System kit, and EconoTaqPLUS Green 2X PCR Master Mix (Lucigen, Middleton, WI, USA). The PCR product was directly cloned into the pCR2.1 vector (Invitrogen Life Technologies, Waltham, MA, USA). The plasmid was amplified and sent for sequencing. For qPCR experiments, cells were cultured to 80–90% confluency and were untreated or treated with IL-1β for 4 h. RNA was collected and purified as described above, and first-strand cDNA was synthesized by SuperScript III as described above. Samples were further analyzed by using FastStart Universal SYBR Green Master (ROX) (Roche Diagnostics, Indianapolis, IN, USA) qPCR reactions. The quantitative mRNA expression was calculated based on the 2
−ΔΔCT method [
20]. GAPDH was selected as the housekeeping gene for normalization; each gene was running along with GAPDH, and the difference between threshold cycles (CT) was designated as ΔCT. ΔΔCT is the difference between their respective controls. PCRs were run with the 7500 Real Time PCR System (96-well format) (Applied Biosystems., Salt Lake City, UT, USA). IL8-Forward: 5′-TCCTGATTTCTGCAGCTCTGT-3′; IL8-Reverse: 5′-AAATTTGGGGTGGAAAGGTT-3′; CCL20-Forward: 5′-GTGCTGCTACTCCACCTCTG-3′; CCL20-Reverse: 5′-CGTGTGAAGCCCACAATAAA-3′; NFKBIA-Forward: 5′-CCCAAGCACCCGGATACAG-3′; NFKBIA-Reverse: 5′-CATAGCTCTCCTCATCCTCACTCTCT-3′.
4.6. Western Analysis
Cells were cultured to 95% confluency, treated or untreated with IL-1β at different times, as indicated in the descriptions of individual experiments and washed with 1X PBS, and pelleted at 5000× g at 4 °C for 4 min. Cell pellets were lysed with RIPA buffer (1X PBS/1% Nonidet p-40/0.5% sodium deoxycholate/0.1% sodium dodecyl sulfate (SDS)). Cellular debris was removed by centrifugation at 15,000× g for 10 min. The amount of protein in the supernatant solution was determined, and samples were heat-treated in 2X SDS sample loading buffer at 100 °C for 5 min. Equal amounts of samples were fractioned by SDS/PAGE and transferred to nitrocellulose membranes. Membranes were blocked in 5% non-fat skim milk powder in PBS for an hour and then probed with primary antibodies, which were visualized with horseradish peroxidase-coupled secondary antibodies by using the Enhanced Chemiluminescence (ECL) Western Blotting Detection System (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA). Human IL-1β (Pepro-Tech., East Windsor, NJ, USA) was used at 10 ng/mL.
4.7. Co-Immunoprecipitation Assay
Cells cultured in 10 cm plates to 95% confluency were lysed in co-immunoprecipitation buffer. Cells were then lysed with co-immunoprecipitation buffer (1% Triton X-100, 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM sodium orthovanadate, 20 μM aprotinin, and 1 mM phenylmethanesulfonyl fluoride and pepstatin A). After spinning the debris for 10 min at 4 °C, the supernatant solution was incubated with EZview Red anti-Flag M2 affinity gel (Sigma-Aldrich, Burlington, MA, USA) overnight at 4 °C. Gel beads were washed with 20 volumes of immunoprecipitation buffer, with rotation at 4 °C for 5 min each time. Protein was eluted with Flag peptide (Sigma-Aldrich, Burlington, MA, USA), following Sigma’s standard protocol. The supernatant solution was mixed with 5XSDS sample loading buffer, boiled for 5 min, and separated in a 10% Tris-HCl SDS/PAGE gel.
4.8. Cell Proliferation and Anchorage-Independent Growth Assays
HT29, DLD1, or HCT116 control cells or cells with overexpression or shRNA knockdown of ARMC4 were plated in triplicates at 20,000/well in a 6-well plate with 3 mL of medium, and the medium was changed every 3 days. Cell number was counted on different days using a cell counting chamber. For anchorage-independent growth assays, type VII agarose (Sigma-Aldrich, Burlington, MA, USA) was autoclaved and mixed with RPMI1640 cell growth medium. Cell culture dishes were coated with 1.2% agarose as the bottom layer. Cells were resuspended in 0.6% of soft agarose and plated on top of the bottom layers. Cells were cultured in the semisolid medium for about 2–3 weeks. The colonies formed were checked under a microscope and measured and counted with the help of ImageJ software (ver. 1.47t).
4.9. Migration Assay
The Boyden chamber containing cell culture inserts with polycarbonate membrane at the bottom with an 8 μM pore size and 6.5 mm diameter (Corning-Costar, Lowell, MA, USA) were used. HT29, DLD1, or HCT116 control cells, or cells with the overexpression or shRNA knockdown of ARMC4 were suspended in serum-free RPMI1640 medium and plated in triplicates at 200,000/insert in the upper chambers. The lower chambers were filled with 10% FBS RPMI1640 medium. The cells were incubated at 37 °C for 72 h. The inserts were then removed, and migratory cells at the bottom of the chamber were fixed and stained in a 4% paraformaldehyde (PFA)/0.1% Crystal Violet solution, followed by washing in deionized water to remove redundant staining. Non-migrated cells remaining at the upper side of the membranes were carefully removed with cotton swabs, and inserts were dried in darkness overnight. The following day stained membranes were pictured in five random non-overlapping fields and counted manually at 20× objective and 20× eyepiece on a transmitted-light microscope.
4.10. Immunohistochemical (IHC) Analysis of Tissue Microarray (TMA)
The colon cancer TMA CO951 (30 cases/95 cores) was purchased from US Biomax, Inc. (Rockville, MD, USA). The cores were duplicated, 30 cancer and 8 of which have matched normal adjacent tissue and 10 cases of matched metastasis, with follow-up data 2 cores per case. All histochemical stains were carried out at the Indiana University School of Medicine IHC Core, using standard procedures included standard deparaffinization in xylene, quenching in 1% hydrogen peroxide/methanol for 10 min, and rehydrated through sequentially graded ethanol. Antigen retrieval was performed by ethylenediamine-tetraacetic acid (EDTA). Using DAKO automated immunostrainers (DAKO, Carpinteria, CA, USA), the slides were blocked for 30 min in horse serum and incubated with ARMC4 antibody, followed by incubation with secondary antibody. The Universal ABC Elite kit (Vectastain, Burlingame, CA, USA) with 3,3′-diaminobenzidine development was used to visualize antibody binding, and the slides were subsequently counterstained with hematoxylin. The tissue arrays were stored at 4 °C and heated to 60 °C for 1 h before use.
4.11. Evaluation of IHC Staining
The TMA sets were scanned using Images were scanned with the Aperio Scanscope Imaging System (Leica Biosystems, Buffalo Grove, IL, USA). The TMA tissue cores were individually copied, labeled, and stored in a separate folder as tiff files. The IHC staining results of individual core tissues were evaluated by an expert pathologist, who was blinded to the patient’s clinocopathological details. The IHC staining was categorized according to a scoring method based on the staining intensity (score 0, no staining intensity; score 1, weak staining intensity; score 2, intermediate staining intensity; and score 3, strong staining intensity). In the case of heterogeneous staining within the samples, the respective higher score was chosen if >50% of the cells showed higher staining intensity.
4.12. Conditioned Media Assay
The HT29, DLD1, and HCT116 cells were seeded into 12-well plates, cultivated to 90% confluency, and transfected with different plasmids: empty vector, ARMC4, shARMC4. After 24 h of transfection, the media were replaced, and the cells were kept for an additional 48 h. The conditioned media were collected, floating cells were pelleted at 3000× g at 4 °C, for 10 min, and the supernatant was aliquoted into sterile tubes and either used immediately or stored at −80 °C. The media were then used to treat 293-NF-κB reporter cells, and luciferase assay was performed as previously described. Detailed procedures have been described by Lu et al., 2009.
4.13. RNA-Seq Analysis
RNA-seq analysis was carried out at the University of Chicago Genomics Facilities. Ultrapure RNAs were prepared essentially described previously [
20]. Briefly, cells were cultured to ~90% confluence, and total RNA was isolated using TRIzol
® reagent. RNA samples were further processed by the University of Chicago Genomic Facilities for the RNA-seq experiment.
4.14. Ingenuity Pathway Analysis (IPA)
Groups of genes were analyzed by the IPA software11. The setting and filter were as follows: reference set: Ingenuity Knowledge Base (Genes_Endogenous Chemicals); Relationship to include: Direct and Indirect; Includes Endogenous Chemicals; Filter Summary: Consider only molecules where species_Human OR Rat OR Mouse. The p values for the enrichment test were calculated using Fisher’s exact test, right-tailed. Log10 (p) was visualized to the left of the p-value. p < 0.05 was considered to be statistically significant.
4.15. Statistical Analysis
The data represent the means ± SEM from at least three separate experiments performed in triplicate. The differences between groups were analyzed using Student’s t-test, and a p-value < 0.05 was considered statistically significant. Statistical analyses were carried out using JMP software (ver. 7.0).



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}